Dear Colleagues and Group Members,
Welcome to my summer update, with my usual selection of highlights for medical device regulatory professionals who are interested in what is going on in Russia and the Eurasian Economic Union:
- Will New GMO Regulations Affect Medical Devices?
On 1st July 2017, rules for the mandatory state registration of genetically modified organisms (GMO) and products containing such organisms came into force in Russia.
Initially introduced by Government Resolution No. 839 (link in Russian) in September 2013, these rules were modified at the end of June 2017 by Resolution No. 770 (link in Russian). The scope of these rules was narrowed down for GMO “intended for release into the environment” only, and at the same time medicinal products and medical devices were excluded from the scope of these rules.
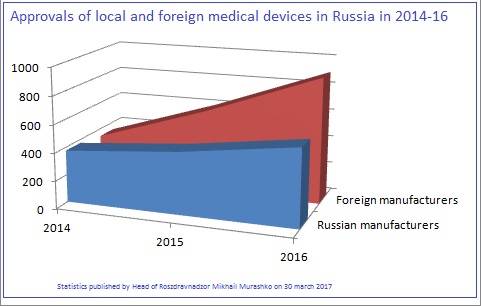
For today, paragraph four of the newly implemented rules underlines that GMO products containing medical devices must be registered within the framework of the established existing medical device registration procedure.
Thus, according to the current legislation for today, medical devices containing GMOs de jure will not require any separate or additional state registration procedure. It is not yet clear, however, if in practice it affects more detailed investigations of medical devices containing genetically modified cells or its products.
GMOs are currently used to produce components of multiple medical products, e.g. recombinant vaccines, clotting factors to treat haemophilia, human growth hormones, recombinant insulin, components of some in-vitro reagents, etc.
- Deadline for Submitting Applications on Prices for Implantable Devices
On 14th July 2017, the deadline passed for medical device manufacturers to submit applications to Roszdravnadzor (Russian healthcare regulator) for the state registration of the maximum selling prices for implantable medical devices.
In this framework, two databases have been recently published on Roszdravnadzor’s website:
- The database with information on average prices for medical products classified according to Russian nomenclature classification (link in Russian).
- State register of maximum sales prices for implantable medical devices included in the list of medical products for the programmes of state guarantees of free medical care to citizens (link in Russian).
The procedure for the regulation of prices for implantable medical devices was approved and has been developed by the Russian government since 2015. According to Resolution No. 1517, the price offered by the manufacturer cannot exceed the weighted average selling price, which is calculated by Roszdravnadzor.
- Eurasian Registration – Requirements for Electronic Submission
On 30th June , the Eurasian Economic Commission published Decision No. 78, “On Requirements for the Electronic Form of Applications and Documents of the Registration Dossier Submitted for Registration of Medical Devices” (link in Russian). The document defines requirements for the structure of the registration file for electronic submission, and rules for the completion of documents in electronic form submitted by the applicant to authorised Bodies of the Member States for registration of medical devices.
As of the end of July 2017, there is still no information that the medical device registration process according to harmonised Eurasian rules is working, whereas most of the Eurasian regulations have been formally enforced.