
Dear colleagues and group members,
Welcome to my usual update on medical device regulations in Russia and countries of the Eurasian Economic Union (EAEU). Here are my three highlights for April 2017:
- Lifting Political Barriers for the Launch of Eurasian Economic Union Medical Device Regulations
On 5 April 2017, the Kyrgyz Republic endorsed a law “On Ratification of the Protocol on Accession of the Republic of Armenia to the Agreement on Common Principles and Rules for the circulation of medical products within the EAEU”. According to the provisions of the Protocol, Armenia officially joins the above Agreement. Until this date, ratification of this Protocol was one of the main political factors hampering the full launch of the unified medical device market and regulatory model for EAEU countries. The law entered into force ten days after the day of its official publication.
In this way all the obstacles to beginning the work of a unified pharmaceutical and medical device market have been lifted and all the second-level EAEU medical device regulations (except the quality management system requirements which are not released by the Eurasian Commission yet) are de jure coming into force for all EAEU member states.
It will be recalled that in recent months official fees for the registration of medical devices according to the new Eurasian procedure were published in Russia, Belarus and Armenia.
- Statistics on Medical Device Registration in Russia
At the end of March 2017, Russian medical device regulator Roszdravnadzor published statistics on registration of medical devices for the year 2016. It was reported that 1,465 new medical devices had been approved (which is around 40% higher than the year before) – among them 559 (38%) products manufactured in Russia and 906 (62%) by foreign manufacturers.
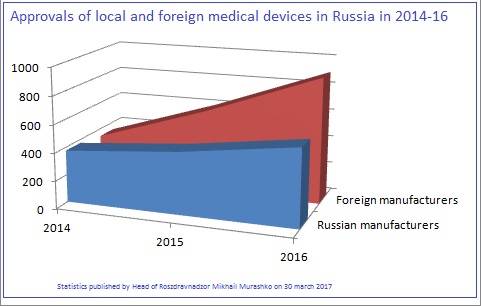
According to the report, the number of registration refusals decreased by around 9% compared to the year before: the regulator reports 477 refusals with a similar ratio: 163 (34%) for local and 314 (66%) for foreign manufacturers. Roszdravnadzor evaluates the total number of registration refusals for the year as 15.2%.
- Quality Management System Inspections in Kazakhstan
On 3 April 2017 the Kazakh medical device regulator (the National Center for Expertise) published a memo (link in Russian) for medical device and pharmaceutical manufacturers clarifying the rules for organising quality management system (QMS) inspection for registration of medical devices and pharmaceutical products. According to the document, for medical device manufacturers QMS inspection is mandatory if a legal manufacturer has never been registered in Kazakhstan before or manufacturing facilities have not been registered in Kazakhstan before, or in cases when conducting an analytical examination is impossible due to the lack or high cost of product samples. The regulator reminds us that the decision about inspection can be taken at any time during the examination. The inspection is carried out within 2-5 working days on one manufacturing site. Valid ISO standard certification of the manufacturer is a necessary condition for the inspection. The Kazak regulator highlights that the manufacturer should: organise the inspection within 30 days after receiving official notification; pay all costs associated with the procedure; and provide translation of necessary information into Russian.
Thank you for following this blog! My objective here is to make Russian and Eurasian medical device regulations clearer. You can also follow my updates on Twitter @MedDevRus
